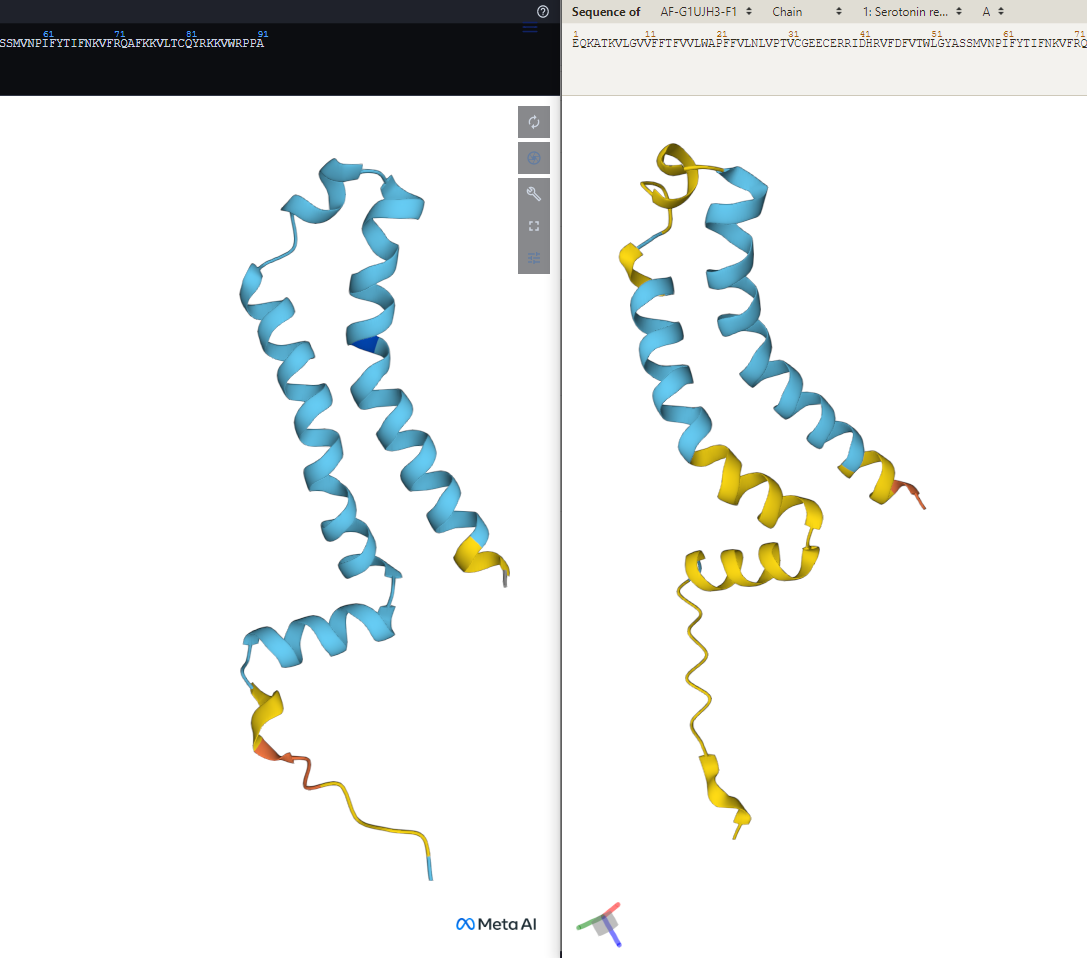
 Shape of a protein predicted by two different AI models (ESMFold on the left, AlphaFold on the right)
Shape of a protein predicted by two different AI models (ESMFold on the left, AlphaFold on the right)cat3cat123 t1_iw6bw1r wrote
Reply to comment by wazabee in Shape of a protein predicted by two different AI models (ESMFold on the left, AlphaFold on the right) by greentea387
To be fair, this modeling is based a lot off of learning from previous crystal structures and the biases those may or may not impose. An x-ray crystal model (or NMR/Cryo-EM structure if you have a very small or very large protein respectively) is still considered the “ground truth”, while these machine learning generated structures are more for hypothesis generation/approximations (that may help in building models from experimental structural data).
Crystal packing needed for x-ray crystallography may rigidify a protein, but at least within the field of structural biology it is not believed to alter the shape of the protein. Likely it just hides the dynamic conformations a protein can occupy - and machine learning methods also suffer from this flaw as they only predict a single structure.
Viewing a single comment thread. View all comments